Computational Chemistry

Beste, Ariana
Brief Bio
Ariana Beste is a research scientist at the Joint Institute for Computational Sciences (JICS). She arrived in the United States in 1999 after earning a B.A. in computer science and completing her M.S. graduate work in chemistry with Professor Gernot Frenking at the Philipps-University in Marburg, Germany. From May to July of that same year, she visited the University of Barcelona, Spain, as a scientist sponsored by the Center Europeu de Parallelism de Barcelona and the Centre de Supercomputació de Catalunya (CEPBA/CESCA). Beste was a Ph.D. graduate student of Professor Rodney J. Bartlett within the Quantum Theory Project of the University of Florida in Gainesville.
She came to ORNL in 2004 as a postdoctoral Fellow with Oak Ridge Associated Universities under advisement of Dr. Robert J. Harrison. Her first appointment as a JICS research scientist was in 2007 in Harrison’s group. She spent 2009 at the Commonwealth Scientific and Industrial Research Organisation in Newcastle, Australia. In 2012, she joined the Computational Chemical and Material Sciences Group, which is led by Dr. Bobby Sumpter. Beste was the recipient of the Richard A. Glenn award from the American Chemical Society, Division of Fuel Chemistry (2011).
Beste has authored or coauthored 26 publications in international journals and three book chapters, and she has given eight invited presentations in local and international meetings.
Research
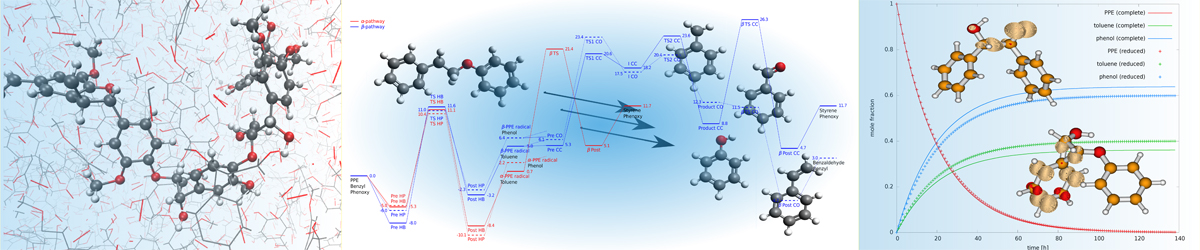
Beste's research interests include the computation of rate constants for complex chemical reaction processes of medium-size systems, applying transition state theory in combination with cost-effective density functional theory. In organic systems with a large number of low-frequency modes, a particular challenge is the correct treatment of anharmonicity. Beste has developed a Python interface to the open-source quantum chemical program package NWChem, in which anharmonic effects are incorporated within the independent mode approximation using the semi-classical Wigner-Kirkwood expansion to compute vibrational partition functions of low-frequency modes. This approach has been successfully applied to study mechanistic details of the thermal degradation of lignin model compounds, including the effect of naturally occurring substituents, and to compute relative rate constant for individual reaction steps. Computed rate constants were further used in kinetic Monte Carlo simulations of lignin model pyrolysis, in which reactant depletion and product formation were followed as a function of time. Computational results were supported by experiments conducted in the group of Dr. A. C. Buchanan, III in the Chemical Sciences division.
Another area of application that Beste has explored in collaboration with Dr. Amit K. Naskar from the Carbon and Composites Group in the Materials Science and Technology division is carbon fiber production from alternative precursors. Kinetic Monte Carlo simulations based on transition state rate constants were used to discriminate between potential reaction paths for the thermolysis of model compounds for sulfonated polyethylene, one of the low-cost precursors studied in Naskar's laboratory. Another promising material for carbon fiber production is lignin. Beste is using reactive force fields as implemented in the LAMMPS program package to obtain a mechanistic understanding of the chemical processes occurring during the oxidation stage.
In collaboration with Dr. Steve H. Overbury from the Surface Chemistry and Catalysis Group in the Chemical Sciences division, Beste is investigating the selectivity of chemical conversion of ethanol on the ceria 111 and 100 surface. Here, she is using density functional methods implemented in the VASP program package, which allows for a periodic treatment of the surfaces. Challenges include the correct localized description of electrons at reduced cerium atoms adjacent to oxygen vacancies and the polar nature of the 100 surface.